


A 510(k) submission is an application submitted to the FDA before a new medical device can be sold. This application shows that the new device is very similar to an existing device that has already been approved by the FDA. In other words, the 510(k) demonstrates that the new device should be just as safe and effective as the existing one.
Simply put, a 510(k) submission is a document package that proves your new medical device is substantially equivalent to an already existing, FDA-cleared device (called a predicate device). This comparison helps establish the safety and effectiveness of your new device without requiring extensive new clinical trials.
510(k) submissions aree important for a few reasons, including:
- Patient Safety: 510(k) submissions ensure new devices meet the same safety and effectiveness standards as existing ones.
- Market Access: A cleared 510(k) allows you to legally market your device in the U.S.
- Faster Approval: Compared to more rigorous premarket approvals, 510(k) submissions offer a potentially faster pathway to market.

There are two main types of 510(k) submissions:
- Traditional 510(k): This requires a detailed comparison of your device to the predicate device across various aspects like design, materials, intended use, and technological characteristics.
- Abbreviated 510(k): This can be used if your device is very similar to the predicate device with only minor modifications. It relies on referencing established standards and guidance documents to demonstrate substantial equivalence.
In addition, the 510(k) submission process involves several steps, including:
- Preparation: This involves gathering necessary documentation, identifying a predicate device, and conducting testing.
- Submission: Submitting the complete 510(k) package to the FDA.
- Review: The FDA reviews your submission for completeness and substantial equivalence.
- Communication: The FDA may request additional information or ask for clarifications.
- Decision: The FDA issues a clearance decision (approval) or a non-clearance decision (denial) with reasons.
This guide will delve deeper into the world of 510(k) submissions. We’ll explore:
- Understanding 510(k) requirements in detail
- Preparing a robust 510(k) submission package
- Common challenges encountered during the process
- Tips and tricks to expedite your 510(k) review

By the end of this guide, you’ll have a comprehensive understanding of the 510(k) process, empowering you to navigate the path to bringing your innovative medical device to market.
Understanding 510(k) Submissions in More Detail
Now that we’ve established the basics of 510(k) submissions, let’s delve deeper into the specifics.
Who’s involved? The Regulatory Landscape
While the FDA is the primary regulatory body governing 510(k) clearances in the United States, other countries may have their own equivalent premarket notification processes. For instance, similar regulations exist in Canada (Health Canada Medical Devices Bureau) and the European Union (CE marking).
Not All Devices Need a 510(k): Eligibility Matters
The 510(k) pathway is generally applicable to Class II medical devices, categorized by the FDA as medium-risk devices. These devices often present a moderate potential for harm but offer significant benefits to patient health.
Here are some examples of Class II devices that might require a 510(k):
- Blood pressure cuffs
- Surgical instruments
- Dental implants
- Powered wheelchairs
Building a Strong Case: Key Components of a 510(k) Submission
A successful 510(k) submission hinges on a well-organized and comprehensive package. Here are some of the essential components:
- Device Description: This section provides a detailed overview of your device, including its design, materials, intended use, and technological characteristics.
- Predicate Device Identification: You’ll need to identify a predicate device, an already FDA-cleared device your device compares to in terms of safety and effectiveness.
- Comparison to Predicate Device: This is the heart of your submission, demonstrating how your device is substantially equivalent to the predicate device. This involves a meticulous comparison across various aspects like:
- Intended Use: Does your device address the same medical condition(s) as the predicate device?
- Technological Characteristics: Are the materials, design principles, and operating mechanisms similar?
- Performance Characteristics: Does your device achieve comparable performance outcomes to the predicate device?
- Risk Analysis: You’ll need to identify potential risks associated with your device and outline mitigation strategies.
- Testing Data: Depending on your device’s complexity and novelty compared to the predicate device, you may need to include data from testing to support your claims of substantial equivalence.
By meticulously compiling this information, you can build a strong case for your device’s safety and effectiveness, paving the way for a smoother 510(k) review process.
Who Submits 510(k) Submissions?
The FDA regulates medical devices to ensure patient safety and effectiveness. When it comes to 510(k) submissions, the responsible party depends on the role they play in bringing the device to market in the U.S. Here’s a breakdown of who typically submits 510(k)s:
- Domestic Manufacturers Introducing a Device: This applies to any U.S. company bringing a new medical device to the U.S. market for the first time.
- Specification Developers: If a company develops the specifications for a device to be manufactured elsewhere but ultimately marketed in the U.S., they are responsible for the 510(k) submission, not the overseas manufacturer.
- Repackagers or Relabelers: Companies that repackage or relabel an existing medical device, especially if the labeling changes significantly affect the device’s use, or their operations introduce new risks, need to submit a 510(k).
- Foreign Manufacturers/Exporters or U.S. Representatives: Foreign companies seeking to introduce their medical device into the U.S. market need to submit a 510(k) themselves. Alternatively, they can appoint a U.S. representative to handle the submission process on their behalf.
In essence, the entity taking the lead in bringing the new medical device to market in the U.S. is typically responsible for submitting the 510(k) application to the FDA.
When Are 510(k) Submissions Required?
Understanding when a 510(k) submission is necessary is crucial for navigating the medical device approval process. Here’s a breakdown of the key scenarios where a 510(k) is mandatory:
Introducing New Devices into Medical Distribution
- First-Time Marketing: This is the most common scenario. Any time a new medical device, not previously marketed in the U.S., is intended for commercial distribution, a 510(k) submission is required.
Following Modifications to Legally Marketed Devices
Even for existing devices, modifications can trigger the need for a new 510(k). This applies when the changes could:
- Significantly Affect Safety or Effectiveness: If the modification alters how the device interacts with the body, its performance characteristics, or introduces new potential risks, a 510(k) is likely necessary.
- Introduce a New Intended Use: If the modification expands the device’s application to address a different medical condition, a new 510(k) may be required to demonstrate safety and effectiveness for the new use.
Remember, the core principle is to ensure the modified device remains substantially equivalent to the original cleared device. The FDA provides a guidance document (https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm) to help determine if a modification necessitates a new 510(k) submission.
While 510(k) submissions are essential for most new medical devices, there are some exceptions. We’ll explore these scenarios in the next section.
Preparing for a 510(k) Submission: Building a Strong Case
A successful 510(k) submission (just like any other regulatory submission, really) hinges on meticulous preparation. This section will guide you through the key steps involved for a successful 510k submission:
A Thorough Regulatory Assessment
Before diving into specifics, it’s crucial to understand the regulatory landscape for your device. A comprehensive assessment will help you determine:
- Classification: The FDA categorizes medical devices into Class I (low risk), Class II (moderate risk), and Class III (high risk). Understanding your device’s class will guide you towards the appropriate regulatory pathway, as 510(k) submissions are typically for Class II devices.
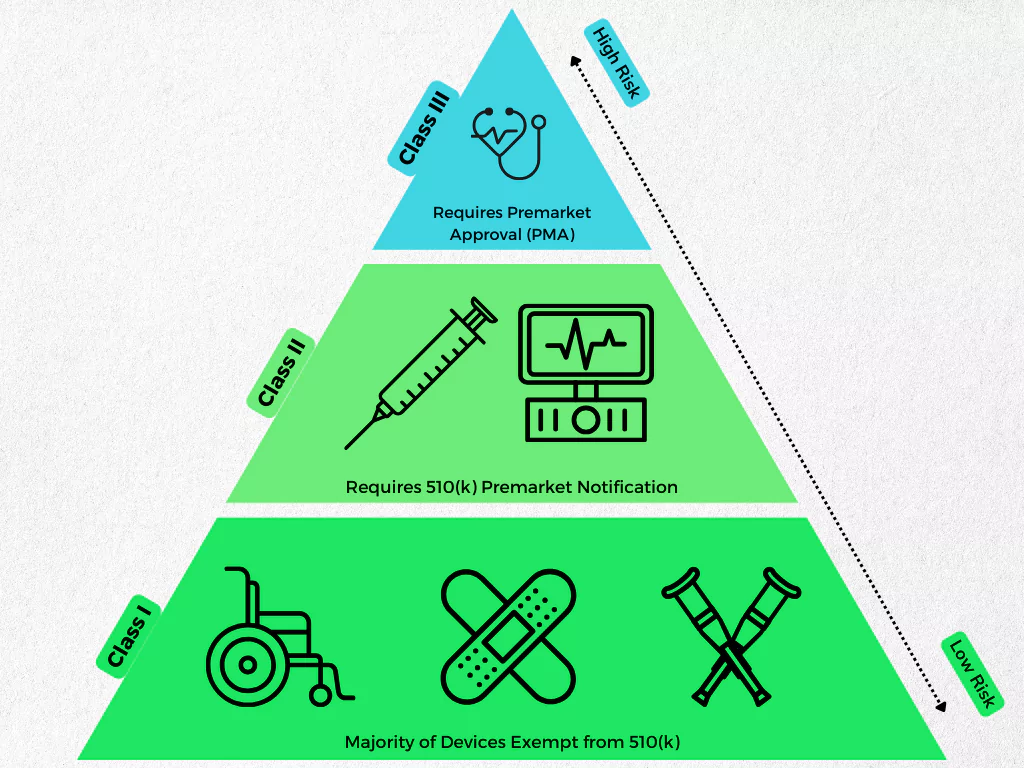
- Exemptions: There may be specific exemptions from the 510(k) requirement for certain Class I or II devices. A thorough assessment will help you identify if your device qualifies for any exemptions.
Identify Predicate Devices and Determine Substantial Equivalence
The concept of substantial equivalence is central to a 510(k) submission. You’ll need to identify an existing, FDA-cleared device (predicate device) that your new device can be compared to in terms of safety and effectiveness. This comparison will form the basis for demonstrating that your device poses an acceptable level of risk and offers comparable benefits.
Here are some factors to consider when selecting a predicate device:
- Intended Use: Does the predicate device address the same medical condition(s) as your device?
- Technological Characteristics: Are the design principles, materials, and operating mechanisms similar?
- Performance Characteristics: Does your device achieve comparable performance outcomes to the predicate device?
Collect and Organize Required Documentation
A well-organized 510(k) submission package demonstrates professionalism and makes FDA’s review process very, very smooth. Here’s a glimpse into some of the key documents you’ll likely need to compile:
- Device Description: This section provides a detailed overview of your device, including its design, materials, intended use, and technological characteristics.
- Predicate Device Identification: Clearly identify the chosen predicate device and provide justification for its selection.
- Comparison to Predicate Device: This is a core component, meticulously comparing your device to the predicate device across various aspects as mentioned previously.
- Risk Analysis: Identify potential risks associated with your device and outline mitigation strategies to ensure patient safety.
- Testing Data: Depending on your device’s complexity and novelty compared to the predicate device, you may need to include data from testing to support your claims of substantial equivalence.
- Labeling: Draft labeling that clearly communicates the device’s intended use, instructions, and safety precautions.
Please note that in most cases, device submissions are sent as 1 large PDF file; containing multiple sections.
Ensure Compliance with Relevant Regulations and Guidelines
The FDA outlines specific regulations and guidance documents that medical device manufacturers must adhere to. For more information, please visit this link to the Overview of Device Regulation on the FDA’s website.
Familiarizing yourself with these guidelines will ensure your submission aligns with the FDA’s expectations. Consulting resources like the Code of Federal Regulations (CFR) and relevant guidance documents from the FDA’s Center for Devices and Radiological Health (CDRH) is recommended.
By following these steps and meticulously preparing your 510(k) submission package, you can increase your chances of a smooth and efficient review process by the FDA.
The 510(k) Submission Process

The 510(k) submission process involves a series of steps, each crucial for obtaining FDA clearance for your medical device. Here’s a simplified breakdown:
- Pre-submission Meetings (Optional): In some cases, pre-submission meetings with the FDA can be helpful to clarify expectations and address any potential concerns before submitting your formal application.
- Compile Application: This involves assembling the complete 510(k) application package, including Form FDA 3514 and all necessary supporting documentation.
- Electronic Submission: Submit the application electronically through the FDA’s Electronic Submission Gateway (ESG).
- FDA Receipt and Tracking: The FDA acknowledges receipt of your submission and assigns a tracking number for reference.
- Completeness Review: The FDA conducts an initial review to ensure your submission is complete and contains all required information.
- Substantive Review: If your submission is complete, the FDA initiates a substantive review, meticulously evaluating your device’s safety and effectiveness based on the 510(k) criteria.
- Additional Information Requests (Optional): The FDA may request additional information (AI) or clarifications if needed to complete their review.
- Applicant Response: You’ll have an opportunity to respond to the FDA’s inquiries and provide any requested information within a specified timeframe.
- FDA Decision: Following a thorough review, the FDA will issue a decision on your 510(k) submission. This can be either clearance (approval) or non-clearance (denial) with reasons explained in a detailed letter.
- Clearance Letter or Non-Clearance Letter: The FDA will formally communicate their decision by issuing a clearance letter or a non-clearance letter to the applicant.
This step-by-step approach hopefully gives you a way forward towards a 510k submission; a successful outcome.
What Common Challenges and Pitfalls Can you Expect During 510(k) Submissions?
Even with meticulous preparation, navigating the 510(k) process presents challenges. Based on various projects, conversations and experience, here are some common pitfalls to be aware of:
- Incomplete or Inadequate Device Description/Performance Data: A clear and comprehensive description of your device and its intended use is paramount. Similarly, providing robust performance data to support your claims of substantial equivalence is crucial. Skimpy details or missing data can raise red flags and delay the review process.
- Predicate Device Selection or Comparison Issues: Choosing the right predicate device and meticulously comparing your device to it forms the core of a 510(k) submission. Selecting an inappropriate predicate device or failing to adequately demonstrate substantial equivalence can lead to rejection.
- Labeling or Instructions for Use Deficiencies: Clear and accurate labeling is essential for safe and effective device use. Ambiguous labeling, missing information, or instructions for use that are difficult to understand can raise concerns about patient safety and potentially derail your submission.
- Regulatory Concerns Identified During Review: The FDA may identify areas where your submission doesn’t meet their established regulations or guidance documents. These could pertain to risk management practices, quality control procedures, or testing protocols. Addressing these concerns promptly and thoroughly is crucial for a successful outcome.
Mitigating these Challenges:
- Early Engagement with Experts: Consulting with regulatory experts or experienced consultants familiar with 510(k) submissions can help you navigate the process effectively and ensure your application is comprehensive and meets FDA expectations.
- Thorough Documentation and Testing: Investing in detailed documentation of your device’s design, development, and testing procedures can strengthen your submission. Likewise, conducting robust testing to generate reliable performance data will bolster your claims of substantial equivalence.
- Staying Current with Regulations: The FDA regularly updates regulations and guidance documents. Staying informed about these changes ensures your submission aligns with the latest requirements.
Speed Up Your 510(k) Submissions: Putting the Pieces Together
Obtaining FDA clearance for your medical device through a 510(k) submission can be a complex but rewarding process. By understanding the key steps and potential challenges, you can navigate this process more strategically and efficiently.
Recap: Key Points for a Successful 510(k) Submission
- Identify the Need for a 510(k): Determine if your device falls under the category that requires a 510(k) submission.
- Prepare Thoroughly: Assemble a comprehensive 510(k) application package with a detailed device description, well-defined comparisons to a predicate device, and robust performance data.
- Ensure Regulatory Compliance: Familiarize yourself with relevant FDA regulations and adhere to established guidelines throughout the development and documentation process.
- Engage with Experts: Consulting with regulatory specialists can provide valuable guidance and ensure your submission aligns with FDA expectations.
- Streamline Your Workflow: Consider utilizing tools and technologies designed to facilitate document management, collaboration, and communication during the 510(k) preparation process.
DocShifter: Automated PDF report generation software for Streamlining 510(k) Submissions
Managing the complex documentation involved in a 510(k) submission can be time-consuming and cumbersome. DocShifter, a powerful document conversion and enrichment software, can help streamline this process.
Whether you need to merge multiple documents into a 510k PDF submission; add bookmarks, hyperlinks, table of contents, header & footers, optimize PDF viewing options, DocShifter significantly speeds up the way you prepare your PDF documents.
Designed to work with any system you have in place today; Ennov, Microsoft SharePoint, OpenText Documentum, Veeva Vault, Generis CARA, LORENZ, file shares, email repositories, and many more.
A well-prepared and efficient 510(k) submission process can significantly expedite FDA clearance and pave the way for the successful launch of your life-changing medical device.
Are you looking to speed up your 510k submissions through document automation? Contact us for your questions or book a free demo.
