

What is a Clinical Trial Application (CTA)?
A Clinical Trial Application (CTA) is a formal request submitted to regulatory authorities to conduct a clinical trial involving human subjects. It’s a comprehensive document outlining the study’s objectives, methodology, risks, and benefits.
Purpose and Importance
The primary purpose of a CTA is to ensure the safety and well-being of human participants involved in a clinical trial. By reviewing the CTA, regulatory authorities assess the scientific merit of the study, the investigator’s qualifications, and the adequacy of the proposed measures to protect participants. It’s a critical step in the drug development process, as it provides a framework for ethical and scientific conduct.
While the fundamental principles of CTA submissions share similarities across jurisdictions, the specific requirements and processes can vary significantly. This article will delve into the core elements of CTA submissions, providing general guidance and highlighting key considerations.
Main Talking Points
- Core components of a CTA
- CTA submission process
- Common challenges and best practices
- Jurisdictional differences (overview)
How Does a CTA Submission Differ Across Jurisdictions?
As mentioned, the terminology surrounding clinical trial applications can vary significantly between different regions. For clarity, let’s break down the key differences:
- United States: In the U.S., the equivalent to a CTA is an Investigational New Drug (IND) application, submitted to the FDA.
- European Union and United Kingdom: In these regions, a CTA is typically submitted alongside an Investigational Medicinal Product Dossier (IMPD). The CTA focuses on the clinical trial protocol, while the IMPD provides comprehensive information about the investigational medicinal product.
- Canada and other regions: In Canada and many other countries, the term “CTA” is used directly to refer to the application for conducting a clinical trial, similar to its use in the EU and UK but without the accompanying IMPD.
Note: While this article provides a general overview of the differences, it’s essential to consult specific regulatory guidelines for each jurisdiction to ensure compliance.
Given the complexities and length that a deep dive into each region would require, we will focus on providing high-level guidance and best practices applicable to CTA submissions in general. In subsequent sections, we will explore the core components of a CTA, the submission process, and common challenges.
EU CTA Submission
CTA Types in the EU
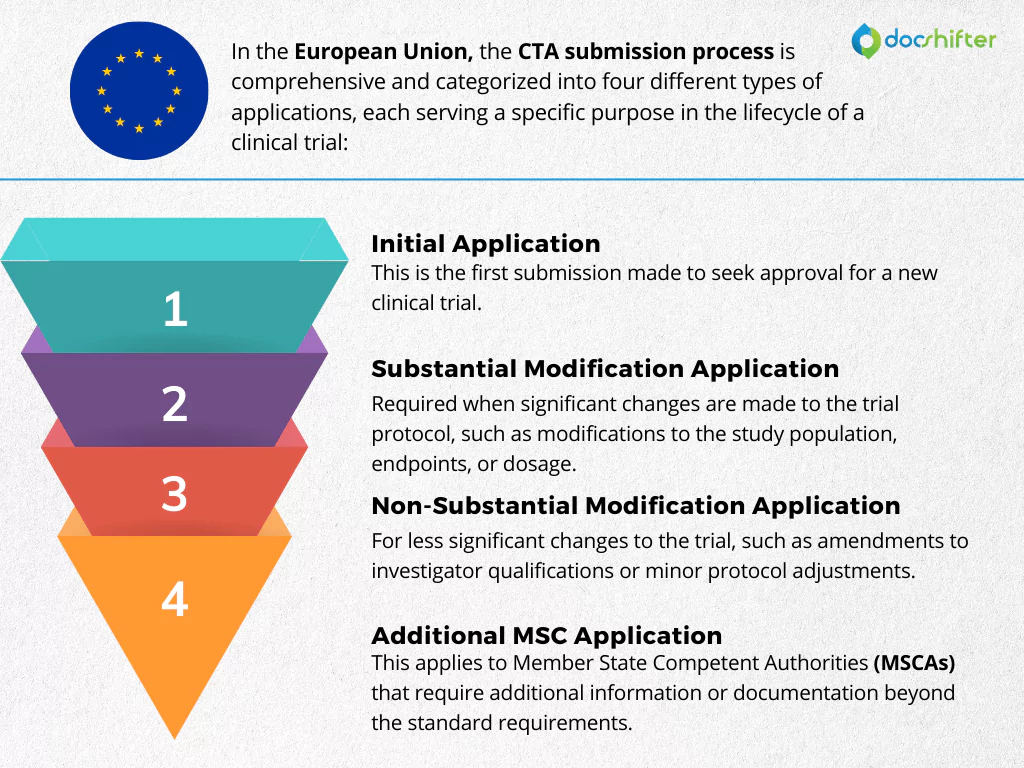
In the European Union, CTAs can be categorized into four primary types:
- Initial Application: This is the first submission for a clinical trial.
- Substantial Modification Application: Required when significant changes are made to the trial protocol, such as modifications to the study population, endpoints, or dosage.
- Non-Substantial Modification Application: For less significant changes to the trial, such as amendments to investigator qualifications or minor protocol adjustments.
- Additional MSC Application: This applies to Member State Competent Authorities (MSCAs) that require additional information or documentation beyond the standard requirements.
Core Components of an EU CTA
While specific requirements can vary between EU member states, a typical CTA generally includes the following essential modules:
| Part I: Scientific and Medicinal Product Documentation | Part II: National and Patient Level Documentation |
| Application form | Patient materials: informed consent, patient information leaflet |
| Protocol | Compensation arrangements |
| Investigators Brochure | Recruitment arrangements |
| GMP documentation | Investigators and facilities suitability |
| IMPD/AMPD | Damage compensation |
| Scientific advice | Data protection rules |
| Pediatric Investigation Plan decision | |
| IMP/auxiliary labels |
UK CTA Submission
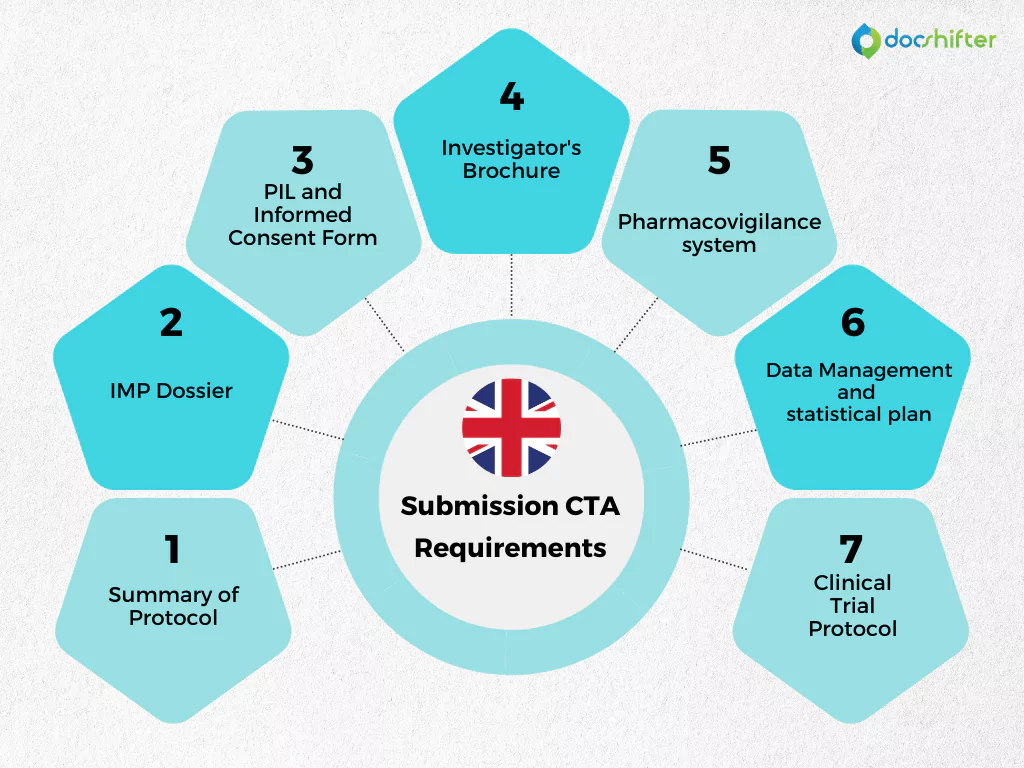
UK CTAs require:
- Summary of protocol
- IMP dossier
- Clinical trial protocol
- PIL and informed consent form
- Investigator’s brochure
- Pharmacovigilance system
- Data management and statistical plan
Key considerations include GCP compliance, ethics committee approval, data protection, and insurance.
Canada CTA Submission
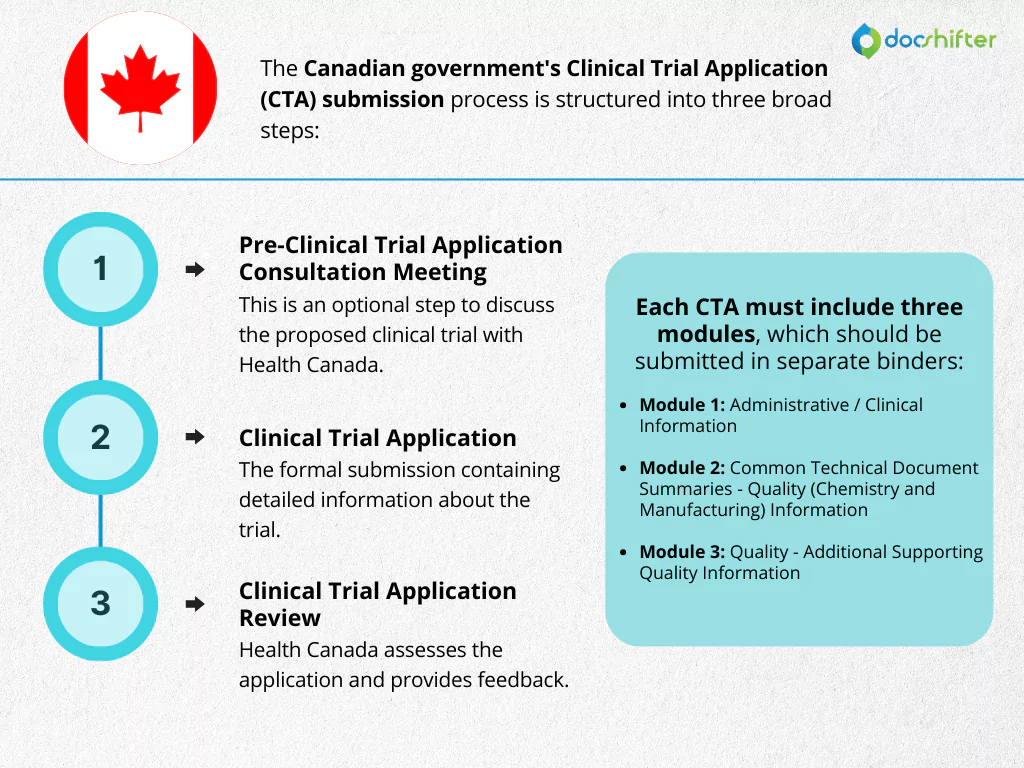
Canada’s CTA submission process involves three primary stages:
- Pre-Clinical Trial Application Consultation Meeting: This is an optional step to discuss the proposed clinical trial with Health Canada.
- Clinical Trial Application: The formal submission containing detailed information about the trial.
- Clinical Trial Application Review: Health Canada assesses the application and provides feedback.
The CTA is divided into three modules:
- Module 1: Administrative / Clinical Information: Includes general information about the trial, study design, and patient population.
- Module 2: Common Technical Document Summaries – Quality (Chemistry and Manufacturing) Information: Contains summaries of the drug substance and product information.
- Module 3: Quality – Additional Supporting Quality Information: Provides detailed quality information as required.
Note: For specific requirements and guidance on each module, please refer to Health Canada’s official webpage about CTA submissions.
CTA Requirements in Other Jurisdictions
While Canada follows a specific process, other countries have their own CTA requirements and submission procedures. To provide a broader perspective, the following table outlines some jurisdictions that have implemented the eCTD 4.0 format for CTA submissions:
| Country | Regulatory Authority |
| United States | FDA |
| European Union | EMA |
| Japan | PMDA |
| Switzerland | Swissmedic |
| Australia | TGA |
It’s crucial to consult the specific guidelines of each regulatory authority for detailed information on CTA requirements and submission procedures.
Preparing for a CTA
EU
Preparing a CTA in the EU involves understanding the different types of applications and their specific requirements. The EMA provides comprehensive guidance on this topic. For detailed information on initial CTA dossiers and submission types, please refer to the following link:
- EMA Guidance on CTAs: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/clinical-trials-human-medicines/clinical-trials-regulation
UK
While specific guidance on preparing for a UK CTA may be limited, it’s essential to adhere to the principles of Good Clinical Practice (GCP) and comply with the UK Medicines and Healthcare products Regulatory Agency (MHRA) regulations. For general information on clinical trials in the UK, you may refer to the MHRA website:
- MHRA Website: https://www.gov.uk/guidance/clinical-trials-for-medicines-apply-for-authorisation-in-the-uk
Canada
Preparing a CTA for submission to Health Canada requires careful attention to the three-module format. For detailed instructions and templates, please consult the following resources:
- Health Canada Guidance: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/clinical-trials/applications.html
Note: It’s crucial to consult the specific guidance documents for the target jurisdiction to ensure compliance with all regulatory requirements.
Putting Together Your Submission
Compiling a comprehensive CTA requires meticulous attention to detail and adherence to specific regulatory guidelines. The following outlines essential components typically included in a CTA submission:
Core Components:
- Protocol: A detailed description of the study design, objectives, methodology, and statistical analysis plan.
- Investigational Medicinal Product (IMP) Dossier: Comprehensive information about the drug substance and product, including quality, safety, and efficacy data.
- Informed Consent Form: A clear and understandable document outlining the trial’s purpose, risks, benefits, and participant rights.
- Investigator’s Brochure: Summary of the available data on the IMP, including preclinical and clinical information.
- Pharmacovigilance System: Describes how adverse events will be monitored and reported.
- Data Management and Statistical Plan: Outlines data collection, management, and analysis procedures.
Note: This list is not exhaustive and may vary depending on the specific requirements of the regulatory authority.
Additional Considerations:
- Ethics Committee Approval: Documentation of ethical review and approval.
- Insurance Coverage: Proof of adequate insurance coverage for trial-related liabilities.
- Patient Information Leaflet: Provides information about the IMP for participants.
- Regulatory Compliance: Adherence to Good Clinical Practice (GCP) guidelines and local regulations.
Working with Health Authorities Post-Submission

Post-submission, sponsors engage in a collaborative process with health authorities (HAs). HAs typically initiate a review period, which involves a comprehensive assessment of the submitted CTA. This evaluation process includes a thorough examination of the application’s completeness and adherence to regulatory standards.
During this phase, effective communication between the sponsor and the HA is paramount. Sponsors may be required to provide additional information or clarify specific aspects of the trial design. The HA will ultimately determine whether the CTA is approved, allowing the trial to proceed, or if revisions are necessary.
Close collaboration with the HA is essential to address any concerns or queries promptly. This proactive approach can expedite the review process and increase the likelihood of a successful outcome.

