

The Food and Drug Administration (FDA) plays a critical role in ensuring the safety and efficacy of drugs entering the U.S. market. One important tool in this process is the Refusal to File (RTF) mechanism.
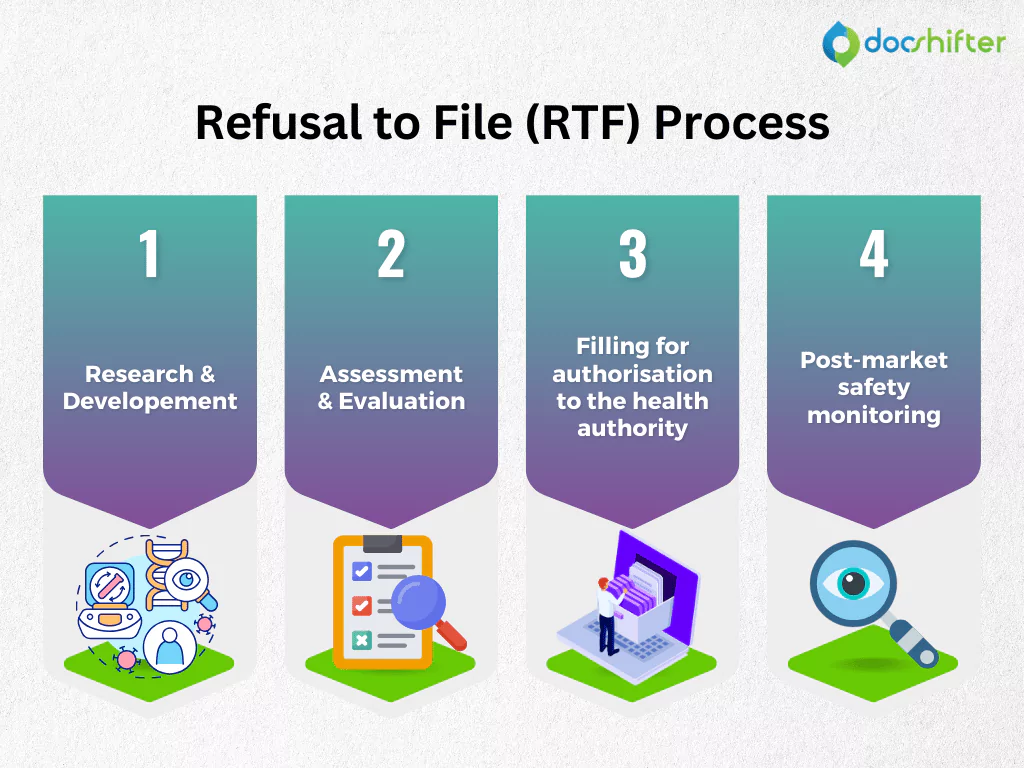
Bringing a new medicine or a product to the market is a complex process that typically takes from 8 to 14 years. Although this process may vary from one Health Authority (HA) to another, there are some general stages that the medicine must go through before it can be safely distributed to patients. It starts with research and development (R&D), then slowly moves into the next steps, including assessment and evaluation through non-clinical and clinical trials, filing for marketing authorization to the HA and getting regulatory approval. The final stage is the post-market safety monitoring after the product is launched to the market.
The RTF letter outlines the specific reasons for the refusal, providing valuable guidance to the sponsor on what needs to be corrected before resubmission. While receiving an RTF can be a setback, it ultimately helps maintain the integrity of the approval process and protects public health.
An RTF is a formal notification from the FDA to a drug sponsor that their application for marketing approval is incomplete and will not be reviewed. This effectively halts the drug’s progress towards approval until the deficiencies are addressed.
What are the consequences of receiving a Refusal to file from the FDA?
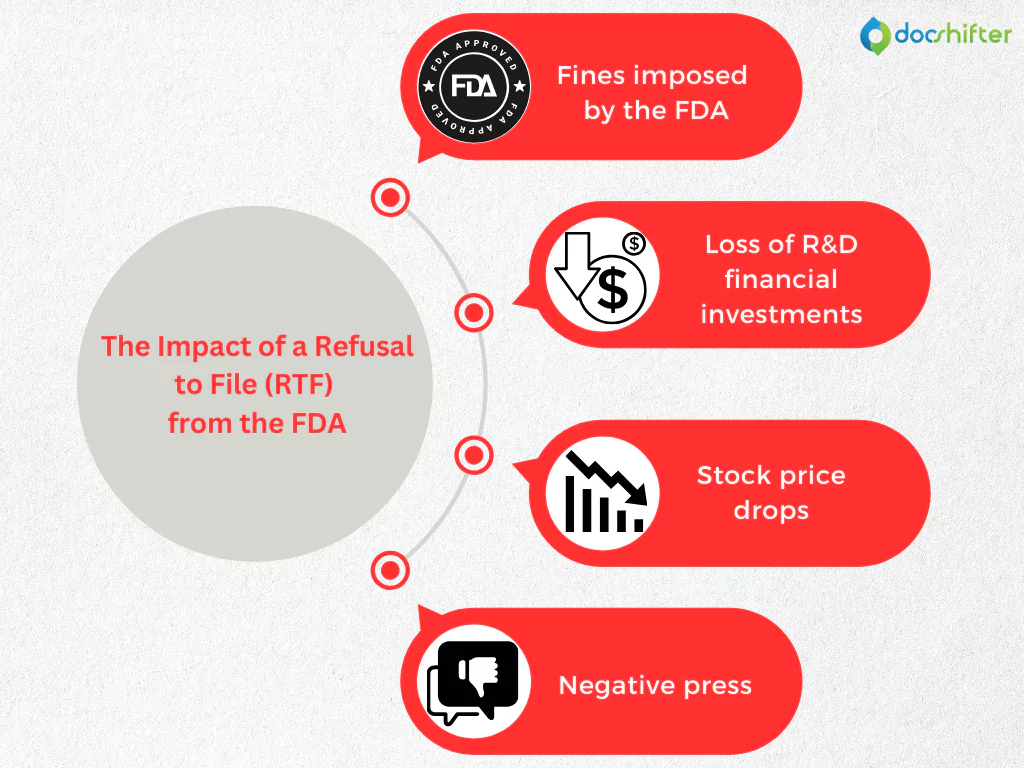
When a Life Sciences organization’s marketing authorization for a new drug is rejected by the HA for non-compliance, the negative impact can be translated into significant financial losses and damages to the company’s image and reputation. To give you a better understanding of the importance of compliance in the Life Sciences industry, let’s focus on the consequences of receiving an RTF from the FDA.
Rejected FDA submissions could cost a life sciences company between $660,000 to $8,000,000 for every day the drug is delayed.
- Fines imposed by the FDA. Under the Prescription Drug User Fee Act (PDUFA), when applying for the approval of a new drug, the pharmaceutical company must pay an application fee (for 2020, the current rates are: $1,471,483 – if no clinical data is required, and $2,942,965 – if clinical data is required). If a drug application is refused for filing, the pharma organization can receive back only 75% of the application fee, incurring financial penalties of $367,871 up to $735,741.
- Loss of R&D financial investments. Statistics in the industry show that pharmaceutical companies spend, on average, 17% of their revenue on developing a new drug. This amount translates into an average of $4 billion, and an RTF letter delays (months) in getting the product to market and causing loss of revenue. Even when the company takes actions to become fully compliant with the FDA’s requirements, it will still lose significant amounts of money every day that it doesn’t bring the new drug to the market.
- Stock price drops. This is another negative impact that contributes to the significant financial losses incurred after receiving an RTF from the FDA. For example, there was the case with a pharmaceutical company that filed for marketing authorization of its new drug with the FDA in December 2015. Two months later, in February 2016, the company announced that it received an RTF letter from the FDA. Soon after this announcement, the stock prices plummeted by 37.3% in a single day.
- Negative press. Ensuring patient safety is vital in the Life Sciences industry. This means that patients also should be confident in the pharmaceutical company’s ability to bring a new drug to the market that may help improve their lives. If the press discovers that the pharma company’s drug application was refused by the FDA, all that negative buzz can end into losing the customers’ confidence. And when that happens, the consequences can be truly catastrophic and it can really be the end of the new drug, long before being launched to the market.
Common Reasons for FDA Refusal to File (RTF)
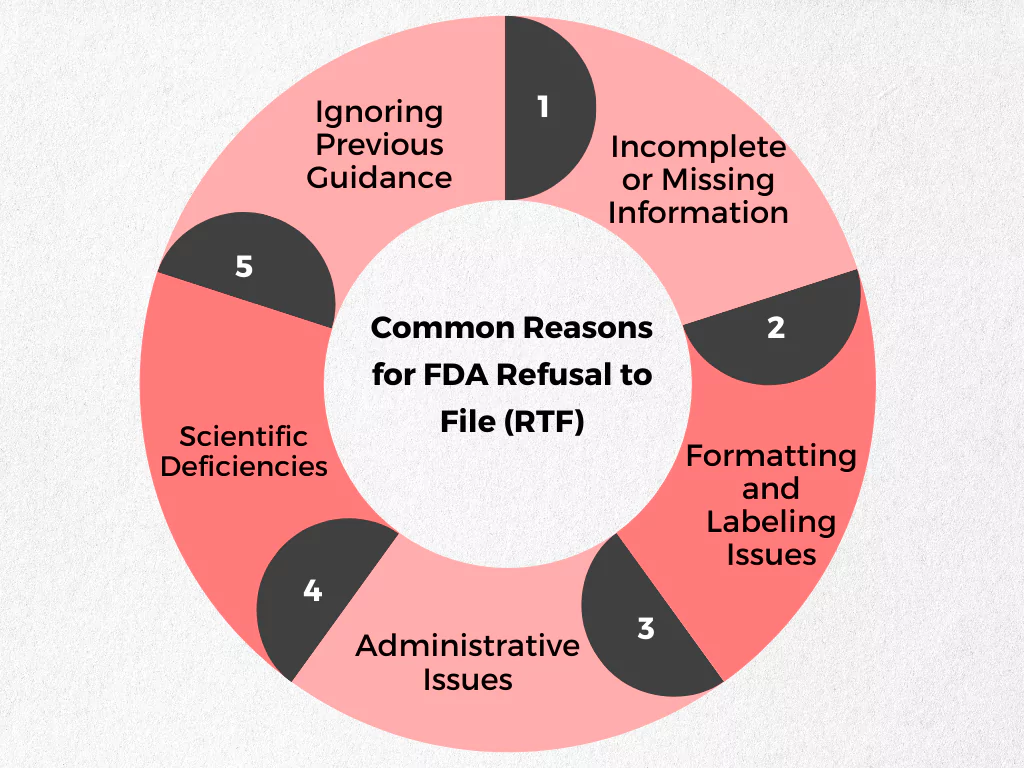
The FDA outlines specific criteria for drug applications to be considered complete and ready for review. Failing to meet these criteria can result in an RTF. Here’s a breakdown of some common reasons for RTFs:
- Incomplete or Missing Information:
- Essential data related to the drug’s safety and efficacy might be missing from the application.
- This could include incomplete clinical trial data, inadequate chemistry, manufacturing, and controls (CMC) information, or insufficient preclinical data.
- Formatting and Labeling Issues:
- The application may not be formatted according to the FDA’s electronic submission standards (eCTD).
- Inaccurate labeling or inconsistencies with the application content could also lead to an RTF.
- Scientific Deficiencies:
- The data presented in the application might not be scientifically sound or raise concerns about the drug’s safety or effectiveness.
- This could involve issues with study design, data analysis, or justification of the proposed dosage.
- Administrative Issues:
- Incomplete or inaccurate application fees, missing required signatures, or failure to comply with communication guidelines set by the FDA could lead to an RTF.
- Ignoring Previous Guidance:
- If a sponsor previously received feedback from the FDA during pre-IND or pre-NDA meetings and fails to address those concerns in their application, an RTF may be issued.
Rejection Process
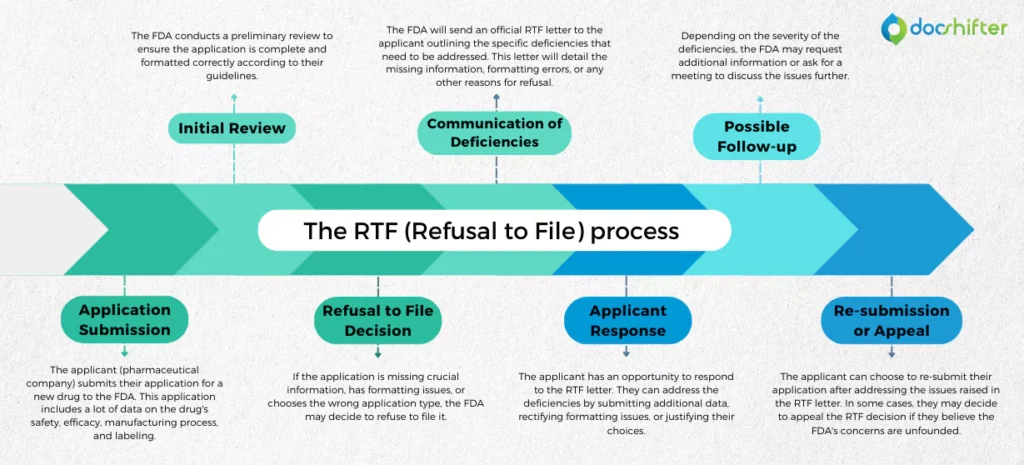
The RTF (Refusal to File) process typically follows these steps:
- Application Submission: The applicant (pharmaceutical company) submits their application for a new drug to the FDA. This application includes a lot of data on the drug’s safety, efficacy, manufacturing process, and labeling.
- Initial Review: The FDA conducts a preliminary review to ensure the application is complete and formatted correctly according to their guidelines.
- Refusal to File Decision: If the application is missing crucial information, has formatting issues, or chooses the wrong application type, the FDA may decide to refuse to file it.
- Communication of Deficiencies: The FDA will send an official RTF letter to the applicant outlining the specific deficiencies that need to be addressed. This letter will detail the missing information, formatting errors, or any other reasons for refusal.
- Applicant Response: The applicant has an opportunity to respond to the RTF letter. They can address the deficiencies by submitting additional data, rectifying formatting issues, or justifying their choices.
- Possible Follow-up: Depending on the severity of the deficiencies, the FDA may request additional information or ask for a meeting to discuss the issues further.
- Re-submission or Appeal: The applicant can choose to re-submit their application after addressing the issues raised in the RTF letter. In some cases, they may decide to appeal the RTF decision if they believe the FDA’s concerns are unfounded.
Important to remember for Refusal to File (RTF)
- The timeframe for the RTF process can vary depending on the complexity of the application and the nature of the deficiencies.
- Communication throughout the process is crucial. The FDA will explain their reasoning for the RTF, and the applicant has the chance to clarify any issues.
- An RTF is not a final decision on the drug’s approval. Once the deficiencies are addressed, the applicant can re-submit their application for a full review.
Achieve technical content compliance with DocShifter
Dealing with compliant content is hard. You need to make sure that everything (and literally everything) is done according to the regulatory requirements. Bookmarks, hyperlinks, table of contents, font requirements, file size limits, image compressions, page margins, and many more.
Whether you deal with Microsoft Word, PDF or any other digital file format, DocShifter software saves you tremendous time in compliant content preparation. Integrated with the leading RIM & eDM systems, DocShifter software supports regulatory teams by:
- Accelerating the drug submission process through automation of renditions for submission-ready PDF documents in eCTD submissions.
- Automating checks and fixes to Word and PDF files to ensure compliance with stringent technical requirements set by health authorities (FDA, EMA, PMDA, Health Canada).
- Automatically merging documents into a single PDF and generating compliant reports for submission and documentation, including 510k and PMA medical device submissions.
- Converting all required digital files for storage in a long-term archive, using PDF/A or the latest file format to ensure digital sustainability for MS Office, image, audio, and video files.
Bonus: We created an FDA PDF format specifications checklist for you, so that you can identify content-related issues as soon as possible to reduce the risk of RTF.
You can download the checklist here.
Did you enjoy this blog post? We have more free knowledge-articles available for you.
Thank you very much for reading our blog post, we hope you enjoyed it.
For more free articles on regulatory topics (eCTD, regulatory submissions, RIM, eCTD v4.0, etc.) we invite you to join our free LinkedIn Regulatory Community.
This community is where we share tips & tricks, updates, and learnings on regulatory topics . The community currently has 2000 members and is growing quickly.
In the first 2 editions, we focused on:
If you think this is interesting, I would like to personally invite you to join the group. And don’t worry, it is completely free.

