

What is an IND submission?
An Investigational New Drug Application (INDA) is a submission made to the Food and Drug Administration (FDA) by a sponsor seeking authorization to conduct clinical trials in humans for a new drug or a new use of an approved drug. The INDA provides detailed information about the drug, the proposed clinical investigation, and the sponsor’s qualifications to conduct the study. Once approved, the IND allows the sponsor to begin clinical testing of the drug in human subjects.
This guide serves as a comprehensive resource for navigating the IND application process. What are the essential steps of an IND submission? How does the review process look like? What do you need to consider to ensure a successful IND application? You will find answers to these questions in this guide.
Table of Contents
- What are the different types of IND Submissions?
- When is an IND submission required?
- Health Authorities for IND Submissions in Select Countries
- The IND Submission Process
- Preparing the IND Application (IND)
- Contents of an IND Submission
- Common Challenges with IND Submissions and How to Overcome Them
- Future Trends in IND Submissions
What are the different types of IND Submissions?
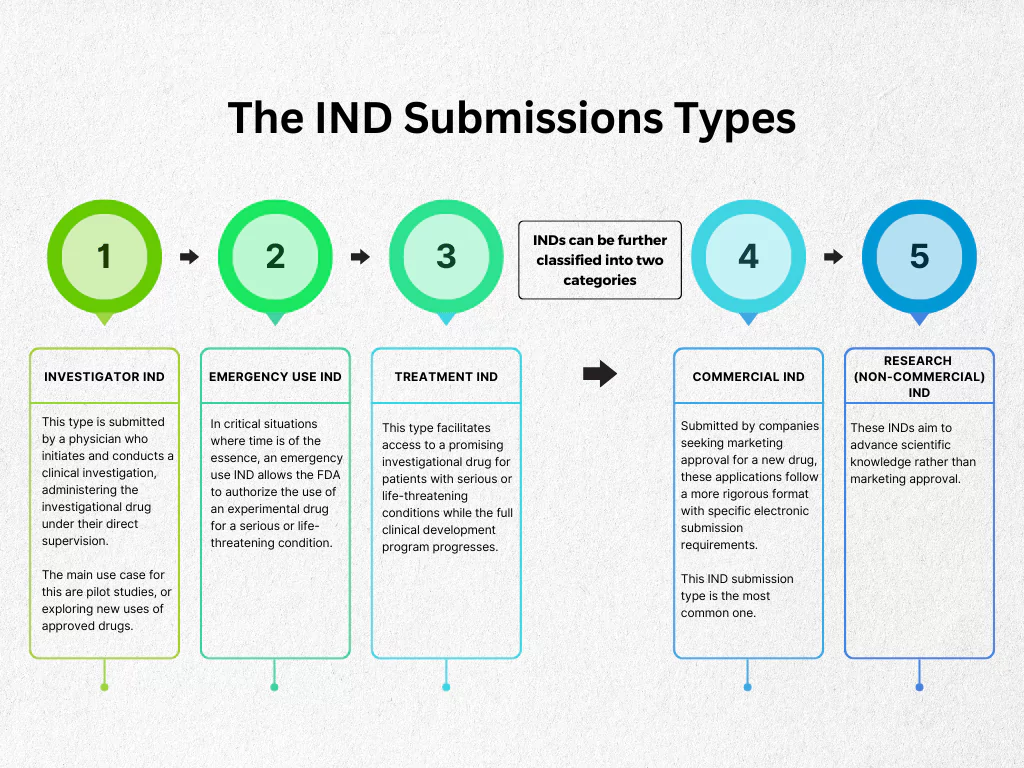
There are three main types of IND submissions, each catering to specific research needs:
- Investigator IND: This type is submitted by a physician who initiates and conducts a clinical investigation, administering the investigational drug under their direct supervision. The main use case for this are pilot studies, or exploring new uses of approved drugs.
- Emergency Use IND: In critical situations where time is of the essence, an emergency use IND allows the FDA to authorize the use of an experimental drug for a serious or life-threatening condition. The most recent example for this is Tecovirimat (TPOXX) for Mpox: In June 2024, the CDC expanded access to Tecovirimat, an antiviral drug, for Mpox patients at high risk of severe complications.
- Treatment IND: This type facilitates access to a promising investigational drug for patients with serious or life-threatening conditions while the full clinical development program progresses.
INDs can be further classified into two categories:
- Commercial IND: Submitted by companies seeking marketing approval for a new drug, these applications follow a more rigorous format with specific electronic submission requirements. This IND submission type is the most common one.
- Research (Non-commercial) IND: These INDs aim to advance scientific knowledge rather than marketing approval.
It would be useful to mention now that while paper submissions are still accepted, many institutions are transitioning to electronic formats for research INDs as well.
When is an IND submission required?
The need for an IND submission depends on whether a drug is intended for investigation in humans. If the study involves animals or solely focuses on developing the drug product itself (e.g., formulation studies), an IND is not required.
The FDA serves as the primary regulatory body for IND submissions in the United States. However, for multinational studies, additional submissions might be necessary to the corresponding health authorities in other countries involved in the trial.
Health Authorities for IND Submissions in Select Countries
| Country | Health Authority | Website |
| United States | Food and Drug Administration (FDA) | https://www.fda.gov/ |
| European Union | European Medicines Agency (EMA) | https://www.ema.europa.eu/en/homepage |
| Canada | Health Canada (HC) | https://www.canada.ca/en/health-canada.html |
| Japan | Pharmaceuticals and Medical Devices Agency (PMDA) | https://www.pmda.go.jp/english/ |
| China | National Medical Products Administration (NMPA) | https://english.nmpa.gov.cn/ |
| Australia | Therapeutic Goods Administration (TGA) | https://www.tga.gov.au/ |
| Brazil | National Health Surveillance Agency (ANVISA) | https://www.gov.br/anvisa/pt-br/english |
Important Note:
- This table provides a selection of some major regulatory bodies. Requirements and submission procedures can vary between countries.
- It’s crucial to consult the specific health authority in each country involved in your multinational study to determine their IND submission requirements.
- Additional regulatory bodies may exist depending on the specific region or country.
The IND Submission Process
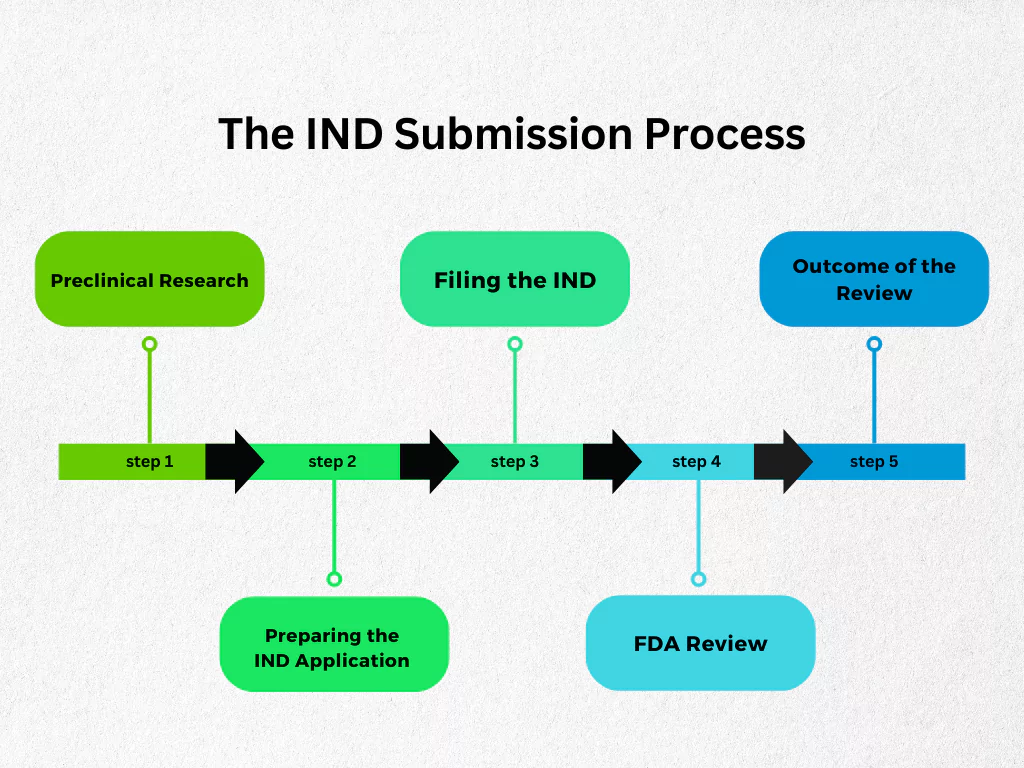
The journey of an IND application can be broken down into several key stages:
Preclinical Research
This foundational stage precedes the IND submission and establishes the scientific rationale for investigating the drug in humans. Preclinical research typically involves:
- In Vitro Studies: Laboratory experiments using cells, tissues, or microorganisms to assess the drug’s potential therapeutic effects and potential for toxicity.
- In Vivo Studies: Animal studies that evaluate the drug’s safety, efficacy, metabolism, and excretion in a living organism. The selection of animal species should be relevant to the intended human use of the drug.
Preparing the IND Application (IND)
The IND application serves as a comprehensive document outlining the proposed clinical investigation and the drug product under study. Here are the core components of an IND, following the FDA’s regulations (21 CFR 312):
- Cover Sheet (Form FDA 1571): This form identifies the sponsor, the drug, and the type of IND submission.
- Table of Contents: Provides a clear roadmap for navigating the large IND document.
- Introductory Statement and General Investigational Plan: This section summarizes the drug, the proposed clinical investigation, and the overall objectives of the study.
- Investigator Brochure (IB): A comprehensive document detailing the drug’s properties, including its chemistry, manufacturing, and controls (CMC) data, pharmacology, toxicology, and any available clinical data. This serves as a crucial reference for investigators involved in the clinical trial.
- Clinical Protocol: A detailed roadmap outlining the study design, methodology, inclusion/exclusion criteria for participants, dosage regimen, assessments, and data collection procedures.
- Investigational Facilities and Investigator Information: This section provides details about the clinical research sites and the qualifications of the investigators who will be conducting the study.
- Chemistry, Manufacturing, and Controls (CMC) Information: Provides data on the drug product’s composition, manufacturing process, quality control procedures, and stability testing results.
- Pharmacology and Toxicology Data: Summarizes the preclinical studies conducted to assess the drug’s safety and potential therapeutic effects.
- Other Pertinent Information: This section may include additional data or documentation deemed relevant to the proposed clinical investigation.
Filing the IND
Once the IND is complete, it is submitted electronically through the FDA’s Electronic Submissions Gateway (ESG) for commercial INDs.
Depending on where the IND submission will be submitted to, INDs can be submitted electronically or on paper. Please check the latest regulations of the health authority you are submitting to.
FDA Review
The FDA meticulously reviews the IND to ensure that the proposed investigation meets the following criteria:
- The risks associated with the study are justified by the potential benefits.
- The rights and safety of human subjects are adequately protected.
- The clinical protocol is scientifically sound and well-designed to yield meaningful data.
The review process typically takes 30 days for a complete IND. However, this timeframe can vary depending on the complexity of the application and the need for additional information from the sponsor.
Outcome of the Review
The FDA communicates the outcome of the IND review through one of three actions:
- Investigational New Drug (IND) Approval: If the application meets all the criteria, the FDA issues an IND approval letter authorizing the sponsor to initiate the clinical investigation.
- Clinical Hold: The FDA may place a clinical hold on the study if there are concerns about the safety of participants, the adequacy of the protocol, or other issues. This essentially pauses the study until the sponsor addresses the concerns and the hold is lifted by the FDA.
- Not Approvable: In rare cases, the FDA may determine that the application is not approvable due to significant deficiencies. They will provide a detailed explanation for this decision, allowing the sponsor to potentially revise and resubmit the IND.
Contents of an IND Submission
As mentioned previously, the core components of an IND application (IND) are outlined in the FDA regulations (21 CFR 312). Here’s a breakdown of some key sections you identified, along with additional elements:
Animal Pharmacology and Toxicology Studies
This section summarizes the preclinical studies conducted to assess the drug’s safety and potential therapeutic effects in animals. It should include:
- Study Design: Describe the type of animal studies conducted (e.g., acute toxicity studies, dose-ranging studies, carcinogenicity studies).
- Methodology: Outline the methods used in the studies, including animal species, dosing regimens, and outcome measures.
- Results: Summarize the key findings from the studies, focusing on the drug’s safety profile, potential therapeutic effects, and any observed side effects.
- Discussion: Interpret the results in the context of the proposed clinical investigation, highlighting how the animal data supports the rationale for testing the drug in humans.
Manufacturing Information
This section provides detailed information on the drug product’s composition, manufacturing process, and quality control measures:
- Drug Substance Information: Describe the active ingredient(s) of the drug, their source, and the manufacturing process.
- Drug Product Information: Outline the formulation of the drug product (e.g., tablets, capsules, injections), including all inactive ingredients and their functions. Explain the manufacturing process for the drug product and the packaging used.
- Quality Control Procedures: Detail the tests and specifications used to ensure the quality, purity, and consistency of the drug product throughout its manufacturing process.
- Stability Testing: Provide data on the stability of the drug product under various storage conditions, demonstrating its shelf life and how it should be stored.
Clinical Protocols and Investigator Information
This section outlines the plan for the clinical investigation and details the qualifications of the personnel involved:
- Clinical Protocol: This comprehensive document details the study design, methodology, inclusion/exclusion criteria for participants, dosage regimen, assessments, and data collection procedures.
- Investigator Brochure (IB): A key reference for investigators, the IB provides detailed information about the drug, including its pharmacology, toxicology, preclinical and any available clinical data, chemistry, manufacturing, and controls (CMC) information.
- Investigational Facilities and Investigator Information: This section provides details about the clinical research sites and the qualifications of the investigators who will be conducting the study. It should include information on the investigators’ experience with the therapeutic area and their training in Good Clinical Practice (GCP).
Additional IND Components
The IND may also include other pertinent information, depending on the specific drug and study design:
- Labeling: Proposed labeling for the investigational drug product.
- Statistical Analysis Plan: A detailed plan outlining how the collected data will be analyzed.
- Informed Consent Documents: The documents used to obtain informed consent from participants in the clinical investigation.
Common Challenges with IND Submissions and How to Overcome Them
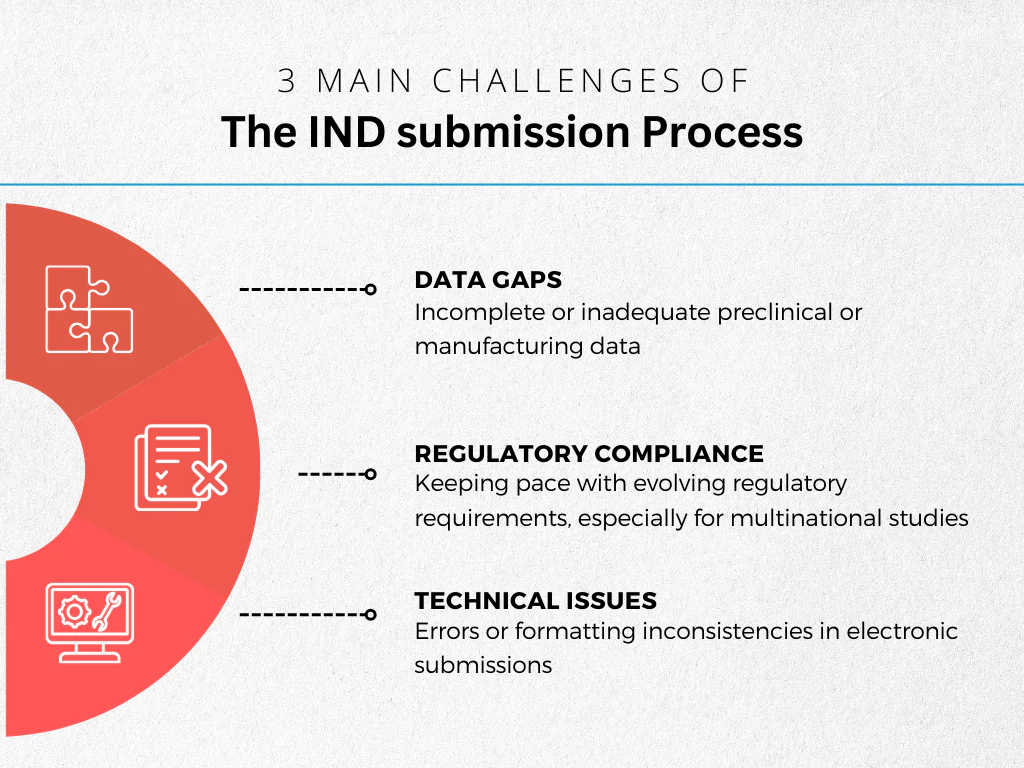
Just like any other submission type, navigating the IND submission process requires careful planning and meticulous attention to detail. Here are some common challenges sponsors may encounter, along with strategies to ensure a smooth application:
Data Gaps
| Challenge | Solution |
| Incomplete or inadequate preclinical or manufacturing data | * Thorough Preclinical Research: Invest in comprehensive preclinical studies that adequately assess the drug’s safety and efficacy profile. |
| * Meticulous Data Collection: Ensure all relevant data is collected and documented during preclinical research and manufacturing processes. | |
| * Early Consultation with the FDA: Consider pre-IND meetings with the FDA to discuss the data requirements for your specific investigational drug. |
Regulatory Compliance
| Challenge | Solution |
| Keeping pace with evolving regulatory requirements, especially for multinational studies | * Dedicated Regulatory Team: Assemble a team with expertise in IND regulations and experience navigating the FDA approval process. |
| * Stay Updated on Regulations: Monitor regulatory updates from the FDA and other relevant health authorities for changes that might impact your IND submission. Resources like subscriptions to regulatory newsletters or attending industry conferences can be helpful. | |
| * Seek Expert Guidance: Consult with regulatory specialists for complex submissions or those involving international trials. Their experience can help ensure your IND adheres to the latest requirements in all relevant countries. |
Technical Issues
| Challenge | Solution |
| Errors or formatting inconsistencies in electronic submissions | * Invest in Quality Software: Utilize software designed to facilitate electronic submissions and ensure compliance with FDA formatting requirements. Look for software features like automated formatting checks and eCTD template compatibility. |
| * Thorough Internal Review: Implement a rigorous internal review process before submission. This should involve checking for errors, formatting inconsistencies, and completeness of the IND application. Utilize multiple reviewers with expertise in both the scientific content and eCTD formatting. | |
| * Focus on Data Integrity: Maintain accurate and complete electronic records throughout the drug development process. This includes implementing data validation procedures and ensuring proper version control to avoid confusion or missing information. |
Future Trends in IND Submissions
The landscape of IND submissions is constantly evolving, with advancements in technology playing a pivotal role in streamlining the process. Here’s a look at how technology is shaping the future of INDs:
Electronic Submissions
The FDA strongly encourages the use of electronic submissions for IND applications. Electronic Common Technical Document (eCTD) format ensures standardized organization and facilitates efficient review by the FDA. This format allows for easier navigation, searching, and reduces the risk of errors associated with paper submissions. For more information on eCTD 4.0 specifically, please visit this content.
The role of Software solutions in streamlining IND submissions
Several software solutions are emerging to assist sponsors in preparing and submitting INDs. These software solutions are designed to make your life easier when dealing with preparing complex and compliant documentation. Here is what DocShifter can mean for your IND submissions:
- Automated document formatting and validation: DocShifter ensures that IND components adhere to the eCTD specifications, minimizing the risk of formatting errors during submission.
- Centralized compliant document formatting: Even though templates are in place to ensure consistency, not everyone uses templates. Regulatory operations end up dealing with formatting documentation at the last minute. DocShifter centralizes all your compliant document processing and formatting; giving you standardized and uniformed submission-ready, high quality documents.
- Are your documents for IND submission residing in multiple systems? If your content resides in multiple systems, you need a software solution that can connect to multiple systems; and seamlessly process documents for submission-readiness. Whether you use Veeva, Microsoft SharePoint, OpenText Documentum, Generis CARA, LORENZ, Ennov or other RIM/DMS systems, DocShifter automation is here to help.
This trend towards leveraging technology can significantly enhance the efficiency and accuracy of IND submissions, ultimately accelerating the development of new drugs. Including a similar section on future trends using technology can definitely be a valuable addition to other submission guidance articles.
Conclusion
The IND submission process plays a critical role in ushering in the next generation of safe and effective drugs. This guide has explored the essential components of an IND, the various stages of the FDA review process, and common challenges sponsors might encounter. By meticulously preparing a comprehensive IND and staying abreast of evolving regulations, sponsors can navigate the process efficiently and contribute to the advancement of life-saving therapies.
Taking the Next Step
Ready to leverage technology to streamline your IND submissions and accelerate your drug development journey?
- Join our Accelerating eCTD Submissions LinkedIn Newsletter: Connect with a community of pharmaceutical professionals and gain access to valuable insights and free tips on INDs, eCTD submissions and lots of other topics.

- Contact us directly and see how compliant document preparation for IND submissions can be much faster and easier.


