

PMA (Premarket Approval) Submissions: Guidance, Process & Requirements
Premarket Approval (PMA) submissions are the cornerstone of obtaining FDA clearance for high-risk Class III medical devices, demonstrating their safety and efficacy through a rigorous regulatory review process.
PMA submissions hold immense importance for medical device manufacturers seeking market authorization for their innovative creations. They ensure patients have access to safe and effective tools for diagnosis and treatment. There are various types of PMA submissions, including traditional, modular, product development protocol (PDP), and humanitarian device exemption (HDE) pathways.
Beyond submission types, it’s crucial to understand factors like PMA application contents, the review process, and post-market requirements.
That’s why we created this guide. This guide should hopefully help you understand what PMA submissions are; and how to prepare yourself for a successful PMA submission. From initial preparation to final FDA approval.
Understanding PMA Submissions in Greater Detail
Following the analogy of our 510(k) submission guide, let’s have a deeper look at PMA submissions. From the regulatory landscape to applicable medical devices and to core components of a PMA package. Everything you need to know about PMA submissions.
Regulatory Bodies Involved in PMA Approvals
Just like the 510(k) article, we will primarily be talking about the US FDA; but will also not neglect the other regulatory bodies in other regions.
The primary regulatory body governing PMA submissions in the United States is the Food and Drug Administration (FDA). However, for international marketing, you may need to navigate the approval processes of other regulatory bodies such as:
- European Medicines Agency (EMA): Responsible for overseeing medicinal products for human and veterinary use within the European Union (EU).
- Health Canada: The regulatory authority for medical devices in Canada.
- Ministry of Health, Labour and Welfare (MHLW): Oversees medical device approvals in Japan.
Table 1: International Regulatory Bodies and PMA Requirements
| Regulatory Body | Location | Website Link | PMA Requirements Link |
| Food and Drug Administration (FDA) | United States | https://www.fda.gov/ | https://www.fda.gov/medical-devices/premarket-approval-pma/pma-application-contents |
| European Medicines Agency (EMA) | European Union | https://www.ema.europa.eu/en/homepage | https://www.ema.europa.eu/en/human-regulatory-overview/medical-devices |
| Health Canada | Canada | https://www.canada.ca/en/health-canada.html | https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices.html |
| Ministry of Health, Labour and Welfare (MHLW) | Japan | https://www.mhlw.go.jp/english/ | https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/index.html (Japanese only) |
Types of Medical Devices Requiring PMA Approval
The FDA outlines specific criteria for determining whether a device falls under the Class III category, necessitating a PMA submission.
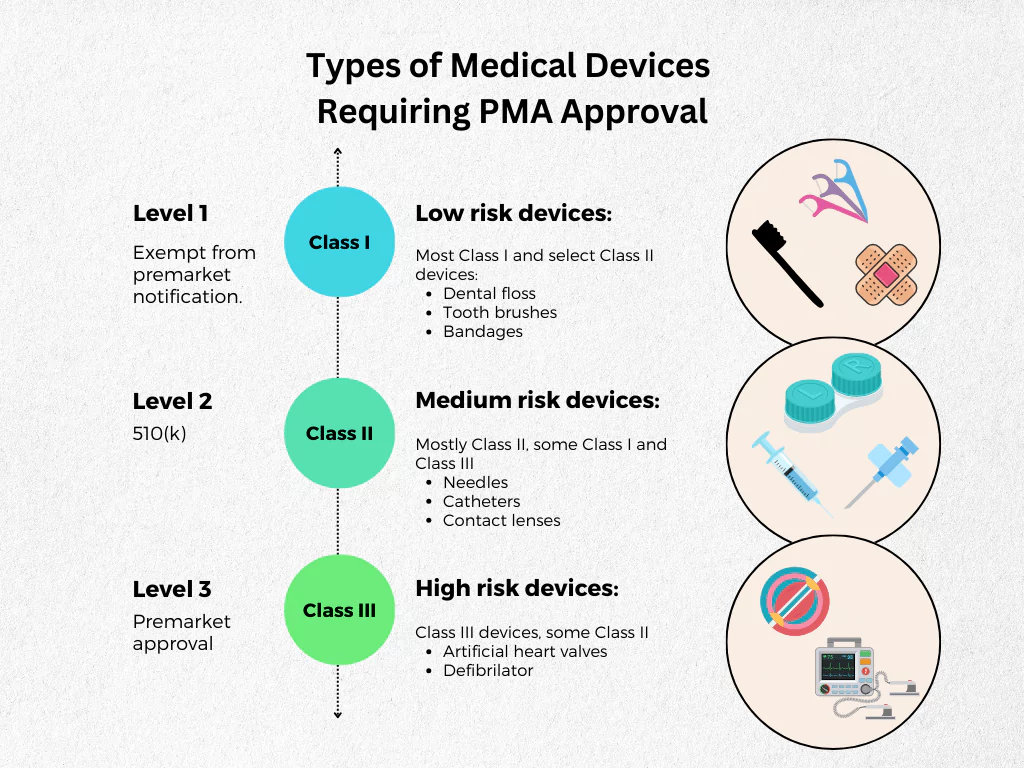
Generally, Class III devices are considered high-risk and present a significant potential for serious risk to the health or safety of users. Examples include:
- Implantable devices:ー pacemakers, artificial joints, and heart valves.
- Life-supporting or life-sustaining devices: ventilators and defibrillators.
- Devices containing new materials or using novel technologies.
Key Components of a PMA Submission Package
A comprehensive PMA submission package serves as a blueprint for the FDA to evaluate your device’s safety and effectiveness. Just like what a regulatory submission should do.
Key components of a PMA submission package typically include:
- Device Description: A detailed description of the device’s design, materials, and intended use.
- Non-Clinical Testing: Data from laboratory testing to demonstrate the device’s functionality and performance.
- Clinical Data: Results from clinical trials demonstrating the device’s safety and effectiveness in humans.
- Quality System: Documentation outlining the manufacturer’s quality control and production processes.
- Labeling: Proposed labeling for the device, including instructions for use and safety information.
Please note that different PMA submission types (including traditional, modular, product development protocol (PDP), and humanitarian device exemption (HDE) pathways might require different components. Please check the corresponding regulatory bodies’ website for the most recent information.
PMA vs. 510(k) Submissions: Is there a difference?
One of the most frequently asked questions. Do I need to submit a PMA or a 510(k) submission?
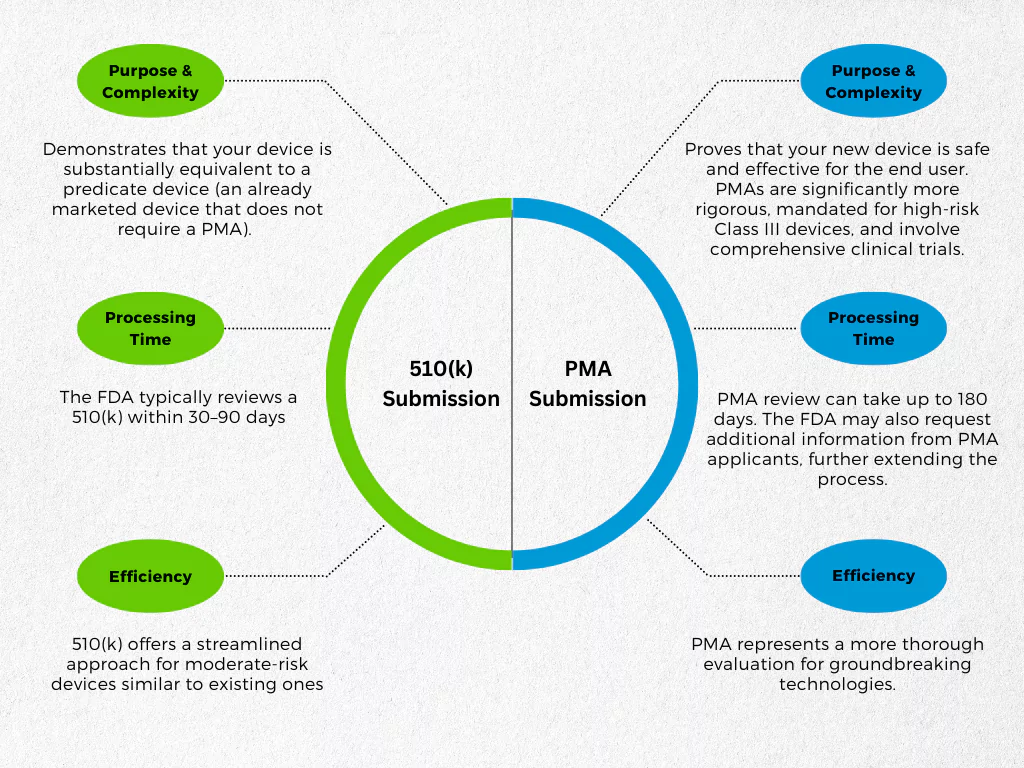
While both PMA and 510(k) submissions secure FDA clearance for medical devices, they are meant to distinct risk categories and involve varying levels of scrutiny. In other words, they are really different from each other.
How are they different? The key differences between PMA & 510(k) submissions lie in their purposes:
- 510(k): Demonstrates that your device is substantially equivalent (as safe and effective) to a predicate device (an already marketed device that does not require a PMA). Think of it as a faster track for devices comparable to existing ones.
- PMA: Proves that your new device is safe and effective for the end user. PMAs are significantly more rigorous, mandated for high-risk Class III devices, and involve comprehensive clinical trials.
This difference in complexity also affects processing times. The FDA typically reviews a 510(k) within 30–90 days, while a PMA review can take up to 180 days. The FDA may also request additional information from PMA applicants, further extending the process.
In essence, 510(k) offers a streamlined approach for moderate-risk devices similar to existing ones, while PMA represents a more thorough evaluation for groundbreaking technologies.
The PMA Submission Process
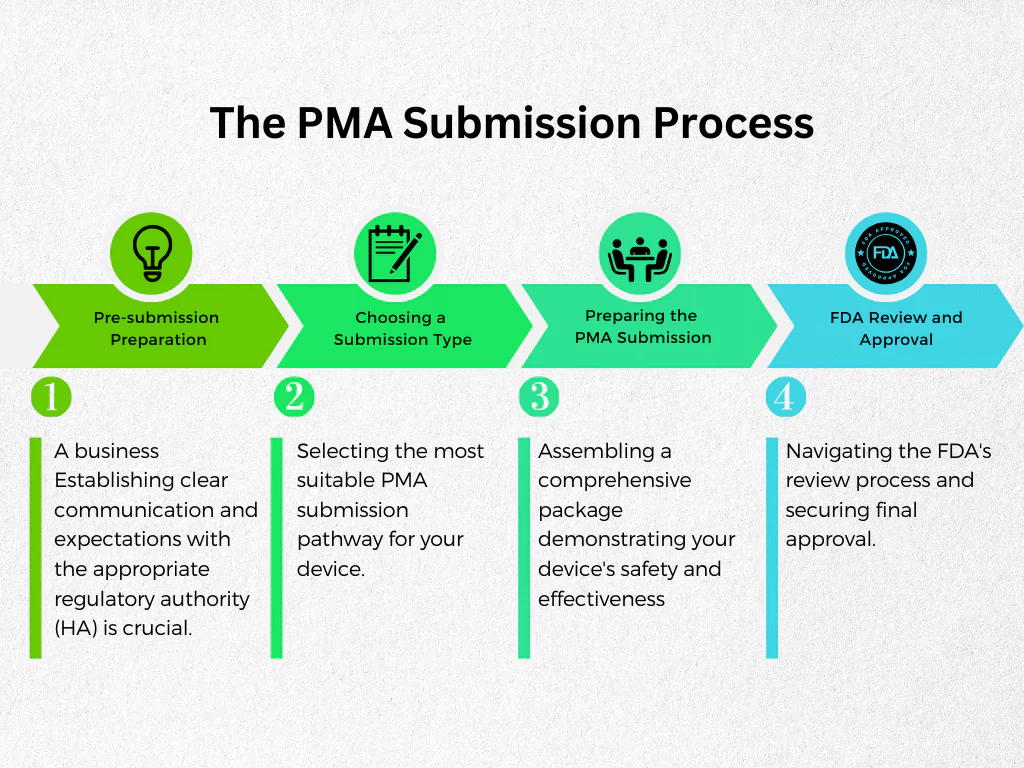
Just like any regulatory submissions, where you not only need to hope but also work hard for a positive outcome, the PMA submission process can be challenging. Needless to say, understanding the key steps involved, you can ensure a smoother and more efficient process. This section will provide a roadmap, guiding you through the essential stages:
- Pre-submission Preparation: Establishing clear communication and expectations with the appropriate regulatory authority (HA) is crucial.
- Choosing a Submission Type: Selecting the most suitable PMA submission pathway for your device.
- Preparing the PMA Submission: Assembling a comprehensive package demonstrating your device’s safety and effectiveness.
- FDA Review and Approval: Navigating the FDA’s review process and securing final approval.
-
Pre-submission Preparation
Engaging in pre-submission meetings with the HA allows you to discuss your device, its intended use, and the planned PMA submission type. These meetings offer valuable insights into the expectations and potential concerns of the regulatory body. During these consultations, you can clarify:
- Data requirements: The specific type and amount of data (clinical trials, non-clinical testing) needed for your submission.
- Device classification: Confirmation of your device’s classification (Class III) and the necessity of a PMA submission.
- Review timelines: Estimated timelines for the HAs review process.
Timelines and Milestones:
The FDA, for instance, outlines a target timeframe of 180 days for reviewing a PMA submission [1]. However, this is an estimate, and the actual review time may vary depending on the complexity of your device and the completeness of your submission. For the most up-to-date information on timelines and milestones, it’s recommended to consult the relevant HA’s website directly. Here are some links to relevant sections on a few key regulatory authority websites:
- FDA – PMA Review Times: https://www.fda.gov/medical-devices/premarket-approval-pma/pma-review-process
It’s important to note that these are just a few examples, and the specific resources will vary depending on the regulatory body you are working with.
-
The 4 Types of PMA Submissions
The FDA offers various PMA submission pathways to accommodate different development approaches. Here’s a brief overview of the four main types:
- Traditional PMA: The most comprehensive pathway, requiring a complete PMA application with all necessary data for a full review.
- Modular PMA: Allows for a staged submission, where modules containing specific data can be submitted and reviewed incrementally.
- Product Development Protocol (PDP): Suitable for devices undergoing continuous development, allowing for iterative submission of data as it becomes available.
- Humanitarian Device Exemption (HDE): An expedited pathway for devices intended to benefit patients with rare diseases or conditions, with relaxed data requirements.
We’ll delve deeper into each of these submission types in the following sections.
-
Preparing for a PMA Submission
Building upon the groundwork laid in pre-submission meetings, this phase involves meticulously assembling a comprehensive PMA submission package. Qualio provides an excellent resource on this topic [link to Qualio guide], particularly their inclusion of a direct link to the FDA’s PMA application form [link to FDA PMA application form on (.gov)]. This form serves as the backbone of your submission, outlining key device details and referencing the supporting data you will provide.
Beyond the core application form, effective PMA preparation encompasses several crucial aspects:
- Conducting a Comprehensive Regulatory Assessment and Strategy Development: A thorough evaluation of your device’s classification, applicable regulatory requirements, and potential challenges is paramount. This assessment will inform your overall PMA strategy, ensuring you gather the necessary data and navigate the process efficiently.
- Designing and Conducting Clinical Trials to Generate Robust Clinical Data: For most PMA submissions, robust clinical trial data is the cornerstone of demonstrating safety and effectiveness. Rigorous clinical trial design and execution, adhering to Good Clinical Practice (GCP) standards, are essential to ensure the validity and reliability of your data.
- Collecting and Organizing Required Documentation: The PMA submission package requires a multitude of documents, including a detailed device description, comprehensive clinical trial protocols, and proposed labeling. Meticulous organization and adherence to formatting guidelines are crucial for a smooth review process.
- Ensuring Compliance with Relevant Regulations and Guidelines: A thorough understanding and implementation of relevant regulations, such as those outlined in the Quality System Regulation (QSR), is mandatory. Additionally, adhering to established guidelines, like those set forth by the International Council for Harmonisation (ICH), demonstrates your commitment to international best practices.
By meticulously addressing these aspects, you can significantly enhance the clarity, completeness, and overall quality of your PMA submission, paving the way for a more streamlined review and approval process.
-
Time for Review: Common Challenges & Pitfalls
After submitting your PMA application, the HA undergoes a rigorous review process to meticulously evaluate your device’s safety and effectiveness. The FDA outlines the four key steps involved in their review process [link to FDA PMA Review Process]:
- Initial Review: Ensures your submission is complete and contains all necessary components.
- Substantive Review: In-depth evaluation of your device’s design, data, and manufacturing processes by relevant FDA experts.
- Panel Review: An optional step where a panel of independent medical and scientific experts may be convened to provide additional insights.
- Decision Announcement: The FDA communicates their final decision (approval, non-approval, or request for additional information).
While the specifics may vary depending on the HA, understanding these general stages can provide a roadmap for navigating the review process.
What to avoid
While the promise of market access awaits, the PMA review process can be fraught with challenges. Here are some common pitfalls organizations encounter:
- Insufficient Clinical Data or Study Design: Robust clinical data is often the cornerstone of a successful PMA submission. Inadequate data quantity or poorly designed studies can raise red flags regarding safety and effectiveness, leading to application rejection.
- Issues with Device Performance, Safety, or Effectiveness: Unforeseen or unresolved issues with the device’s performance, safety profile, or effectiveness during the review process can significantly delay or even derail your application.
- Challenges in Patient Recruitment and Retention for Clinical Trials: Recruiting and retaining a sufficient number of qualified participants for clinical trials can be a significant hurdle, especially for rare diseases or devices targeting specific patient populations. Our suite of clinical trial management tools can streamline this process, facilitating efficient recruitment and data collection.
- Regulatory Concerns or Deficiencies Identified During Review: Inconsistencies with relevant regulations or deficiencies in your manufacturing quality system can raise concerns during the review. Our regulatory compliance expertise can help you navigate complex regulations and ensure your manufacturing processes meet the highest quality standards.
By proactively addressing these potential challenges and leveraging our expertise in clinical trial management and regulatory compliance, you can significantly increase your chances of a smooth and successful PMA review.
Speeding Up Your PMA Submissions & Getting Devices to Market Quicker
One of the ways to get your devices to market quicker, is to take care of the documentation process.

For medical devices, in most cases, regulatory bodies require a large PDF file containing all the study and other data. This PDF file can range anywhere from 100 to thousands of pages; and is typically created by merging various file types together; Microsoft Word, Excel, images, scanned PDFs, etc.
This PDF file needs to be viewed, bookmarked, created, and handled in a compliant way. This way, the FDA or the other regulatory bodies can easily review the PDF.
DocShifter’s automated report level publishing solution automatically merges documents into a single compliant PDF; ensuring correct bookmarks, view settings, PDF options, font settings; and everything else that your PDF needs to be compliant with the regulatory bodies’ requirements.

